Introduction
Registering medical devices with ANVISA is a fundamental requirement to ensure the technical compliance and safety of products available in Brazil. Companies that manufacture or import these products need to understand how to navigate ANVISA’s requirements to obtain approval. This detailed guide explores all stages of the registration process, using flowcharts to aid understanding.
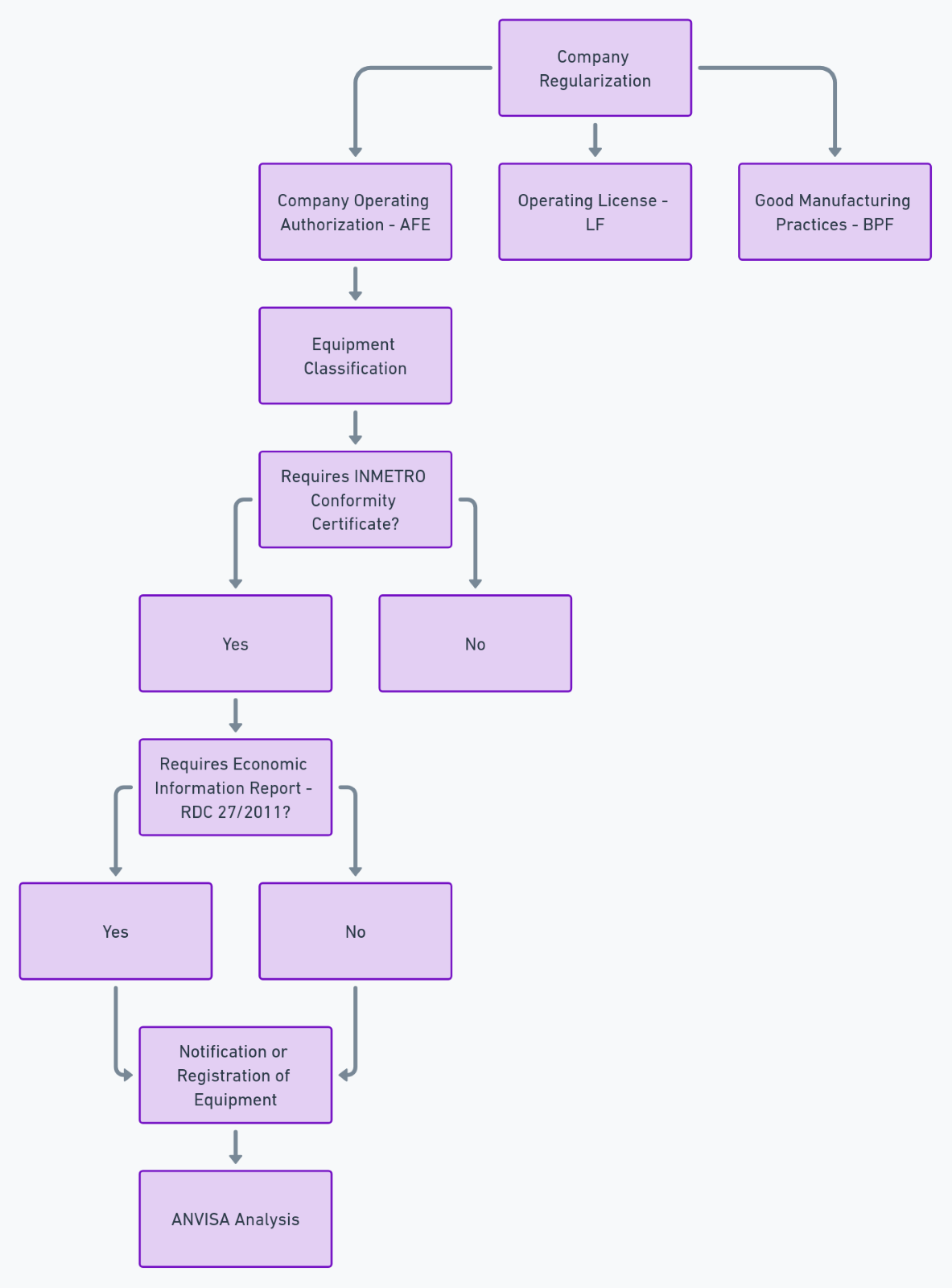
Step 1: Company Regularization
Before starting the registration process, the company must ensure it is properly registered with the health authorities.
1.1 Company Operation Authorization (AFE)
Issued by ANVISA, the AFE is the first document the company must obtain. Only companies established in Brazil can apply for this authorization, and in the case of imported products, the importing company assumes this responsibility.
1.2 Local Operating License (LF)
Issued by local (state or municipal) Health Surveillance, the LF is a mandatory requirement for the company to operate. This document ensures that the company meets the necessary sanitary conditions for manufacturing, importing, or distributing medical products.
1.3 Good Manufacturing Practices Certification (BPF)
This certificate is mandatory for manufacturers and ensures that the company follows strict quality standards in the manufacturing process. Certification may involve local inspections by ANVISA or the municipal Health Surveillance.
1.4 Good Distribution and/or Storage Practices Certification (CBPDA)
The Good Distribution and/or Storage Practices Certification (CBPDA) is issued by ANVISA to certify that an establishment complies with the Good Distribution and Storage Practices or Good Storage Practices as provided for in current legislation. It applies to companies storing, distributing, and importing Medicines, Health Products, and Pharmaceutical Inputs located in the national territory, as defined in specific regulations.
Step 2: Product Sanitary Classification
At this stage, the medical device is classified according to the level of risk it poses to health, ranging from Class I (low risk) to Class IV (maximum risk). The classification defines the technical requirements that need to be met.
- Class I: Low-risk products.
- Class II: Medium-risk products.
- Class III: High-risk products.
- Class IV: Maximum-risk products.
(For more information about risk classification, access our article Notification and Registration of Medical Devices: Differences, Classifications, and the Importance of Regulatory Compliance)
2.1 Identification of Petition Type
Based on the risk classification, the company must determine whether the device is subject to registration or notification:
- Notification: For Class I and II products.
- Registration: Required for Class III and IV products.
Step 3: Electronic Submission
The submission process is done through ANVISA’s electronic system. This service allows the company to formalize its request with ANVISA and obtain information through specific forms. It also enables the generation of the Federal Collection Document (GRU) for paying the Health Surveillance Inspection Fee (TFVS). The company must access the portal and fill in the required information according to the petition type (registration or notification).
3.1 Generate Federal Collection Document (GRU)
At the end of the electronic submission, the company must generate the GRU, which will serve as proof of payment of the inspection fee. The submission will only be considered valid after payment.
Electronic Access Page for Company Registration and Petition Submission:
https://www.gov.br/anvisa/pt-br/setorregulado/administrativo
- Step-by-step information guides for each of these procedures are available on the indicated webpage.
- To identify the petition’s Generating Factor, check the list available on the electronic page:
https://www9.anvisa.gov.br/peticionamento/sat/Consultas/ConsultaAssunto.asp
- Choose “Health Products” in the “Area Selection” field.
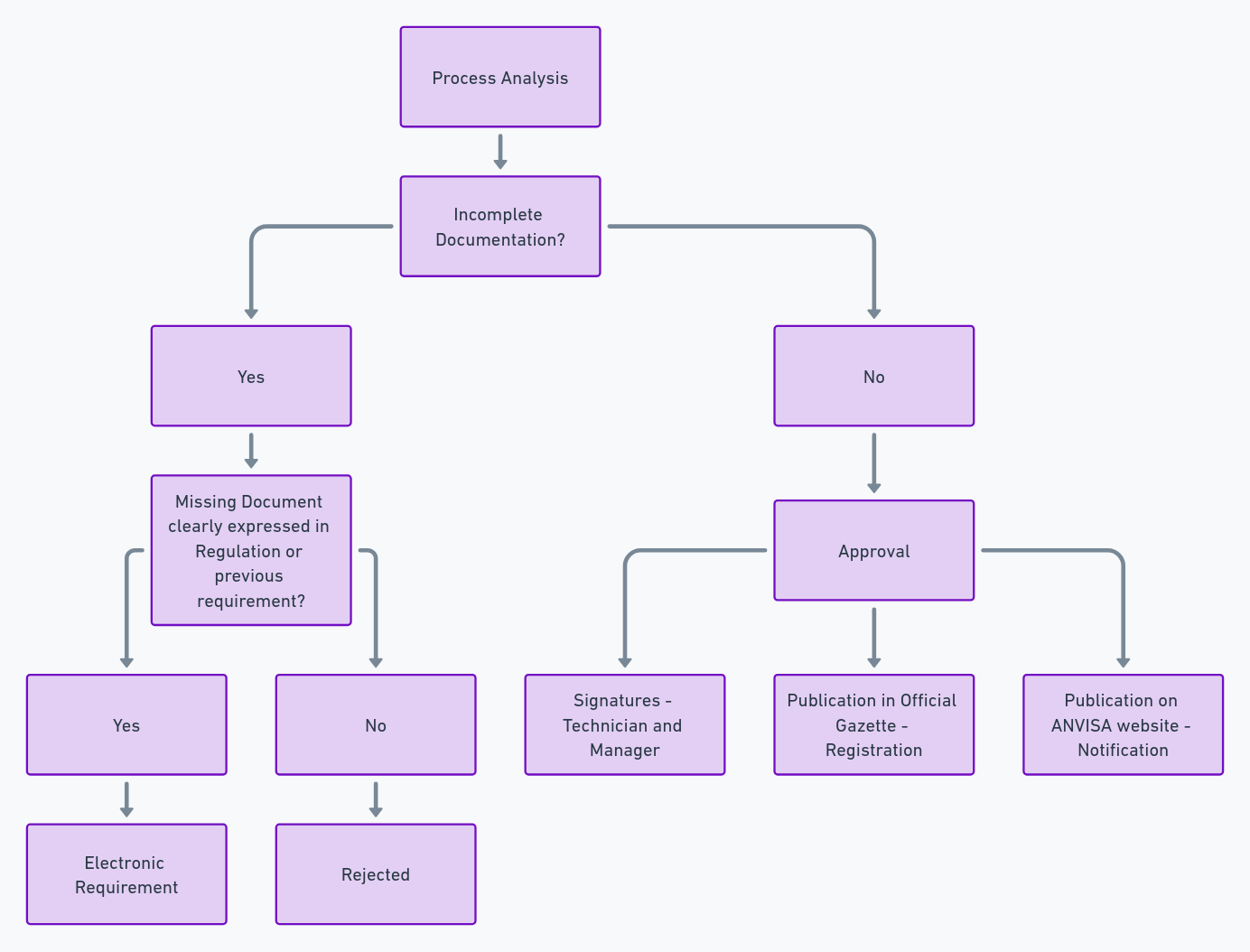
Step 4: Submission and Process Review
4.1 Submission
The documentation must be submitted according to the current procedure established by ANVISA’s Documentation Management. In-person service can be performed at ANVISA’s facilities for clarifications regarding the submission procedures. More information on in-person service and submission procedures is available on ANVISA’s website.
It is essential that the petition documents, when submitted, are duly signed by the Legal Representative and the company’s Technical Officer, as required by Article 6 of Law No. 9,784, of January 29, 1999. All documents related to the product (instructions for use, technical report, petition form, label models, etc.) must bear these signatures.
The submitted petition receives a unique number, consisting of 11 digits, followed by the year of submission and ending with a check digit; the full number is called the “protocol number” (example of a protocol number for a petition submitted in 2021: 25352.XXXXXX/2021-YY), which will be converted into a process number + case number (primary process or primary petition) or case number (secondary petition). The company
should monitor the process through the protocol number, process number, or case number on the website.
https://consultas.anvisa.gov.br/#/documentos
Be aware that the protocol number does not correspond to the process or case number, as these are generated only after the petition is entered into ANVISA’s computerized system.
It is suggested to check the status the day after the submission. When the company needs information about the process or a specific petition, it should always refer to the process number or case number.
4.2 Technical Review
The process review will be conducted by the Medical Equipment Technology Management – GQUIP. During the review, it is checked whether the documents presented in the registration or notification petition comply with the current health legislation. After review, ANVISA may:
- Approve the process: When all requirements are met.
- Issue a Technical Requirement: Requesting additional information or corrections.
- Deny the request: If there is missing documentation or serious technical issues.
Conclusion
After review and publication in the Federal Official Gazette, the medical device will be authorized for commercialization in Brazil. The registration process may seem challenging, but with an understanding of the steps and good document organization, it is possible to expedite approval.
📑 Learn More – Do you want to understand in detail how the process of registering medical devices with ANVISA can benefit your company and ensure consumer safety? For more information about medical device certification, get in touch with Brisa’s experts. Our team is ready to answer your questions and guide you through all stages of the certification process, ensuring compliance and efficiency.
Access now and stay informed!
Find out more about BPO in RA!
*Budget for registration ownership transfer, Market Access Strategy, and BPO in RA services for your company: www.brisa.com.br
